Le site de LASE a déménagé. Il se trouve maintenant ŕ
l'adresse http://xlase.free.fr/. Vous trouverez
ci-dessous l'ancienne version de la page.
Qu'est-ce que LASE ?
LASE est un logiciel dédié à l'analyse des
spectres d'absorption des rayons X. Il tourne sur toute machine
disposant soit d'unix et X-Window, soit de Windows (95 ou 98), soit
du TOS ou compatible (Magic Mac par exemple). Dans les trois cas, il
dispose d'une interface graphique conviviale, qui permet de
visualiser en temps réel l'effet des différents
paramètres tout en laissant à l'utilisateur une grande
liberté de choix.
LASE a été conçu au LURE, dans le cadre de
l'analyse par spectroscopie d'absorption X de composés
bioinorganiques. Il est donc particulièrement adapté
à l'étude de tels composés :
protéines, complexes de métaux de transition,...
LASE dispose d'options pour le traitement et l'analyse des
spectres dans trois domaines :
Ces différents points sont repris et
détaillés dans la suite de ce document.
Comment obtenir LASE ?
Cela dépend de la configuration que vous utilisez. Dans
tous les cas, merci de mentionner l'utilisation de LASE lorsque vous
présentez des résultats obtenus à l'aide de
LASE.
Télécharger LASE
Dates de mise ŕ jour
Dates de derničre mise ŕ jour
| Version | format zip | format gzip | format bzip2 |
| X-Window | 25 mai 2001 | 25 mai 2001 | 25 mai 2001 |
| Windows | 23 janvier 2001 | | / |
| TOS | 6 février 2000 | / | / |
| Exemples | | | / |
| Documentation | 17 avril 2000 | 17 avril 2000 | 17 avril 2000 |
Attention ! La version Windows est encore
expérimentale. Certaines fonctions ne sont pas disponibles, d'autres ne sont
pas entičrement implémentées. La version X-Window est nettement préférable
pour un usage courant.
Pour pouvoir profiter de la visualisation tridimensionnelle du
modèle structural, vous devez disposer d'une
bibliothèque 3D compatible Open-GL. Voyez par exemple
Mesa.
Pour connaître la liste des nouveautés depuis la derničre version, voyez ici
Extraction des spectres EXAFS
LASE dispose de nombreuses options permettant de traiter les
données expérimentales brutes pour en extraire le
signal EXAFS. Il inclut en particulier des outils statistiques
permettant de gérer l'information issue de plusieurs
enregistrements d'un même spectre.
Récupération des données
expérimentales
- Gestion des spectres enregistrés en transmission, en
fluorescence ou en détection d'électrons (fichiers
expérimentaux du LURE
préférentiellement) ;
- Possibilité de garder I0, I1 et
de recalculer le coefficient d'absorption avec des I0
ou I1 modifiés ;
- Gestion des expériences avec un détecteur
multi-éléments : chargement des
éléments séparément ou avec moyenne
automatique, choix des éléments à utiliser,
normalisation automatique ;
- Gestion des expériences en transmission avec trois
chambres d'ionisation (pour la calibration).
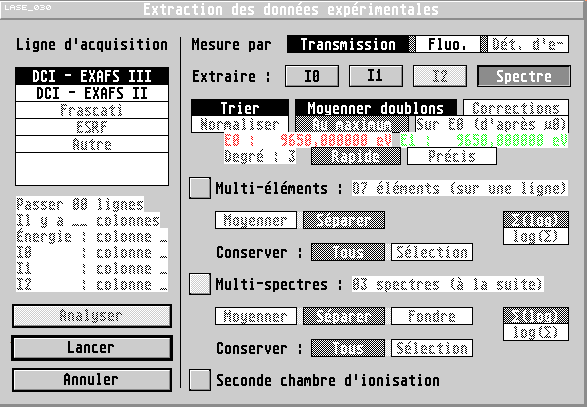
Traitement d'ensemble du spectre
- Recalibration, linéaire ou avec utilisation de la loi de Bragg ;
- Dérivation, intégration, interpolation, troncature,... ;
- Élimination des glitches par interpolation (polynômiale ou par spline
cubique) ;
- Normalisation, avec 1 ou 2 points ou par modélisation du
µ0 pour trouver le seuil ;
- Recherche de l'énergie de seuil (maximum de la dérivée) ;
- Tri, recherche et traitement des doublons suivant plusieurs critčres.
Analyse statistique des spectres
- Calcul du spectre moyen de n spectres, avec écart-type pour
chaque point du spectre moyen ;
- Visualisation facultative des barres d'erreur point par point
pour un spectre ;
- Toutes les erreurs sont propagées lors de l'extraction du signal ;
- Les erreurs peuvent ętre propagées lors du filtrage par transformation
de Fourier, avec calcul de la matrice de covariance ;
- Outils statistiques de base : moyenne, variance ;
analyse de distribution (histogrammes, test de Kolmogorov-Smirnov
pour une distribution gaussienne)
Extraction du signal EXAFS
- À chaque étape, le résultat est
affiché en temps réel après toute
modification d'un paramètre ;
- Absorption de la matrice modélisée par une
victorine, complète ou tronquée, ou un
polynôme &emdash; calculés par les moindres
carrés sur le préseuil dans une région
définie par l'utilisateur ;

- Élimination du µ0 en trois
étapes : polynôme en E, polynôme en
k, spline contrainte &emdash; tout étant
contrôlable par l'utilisateur ;
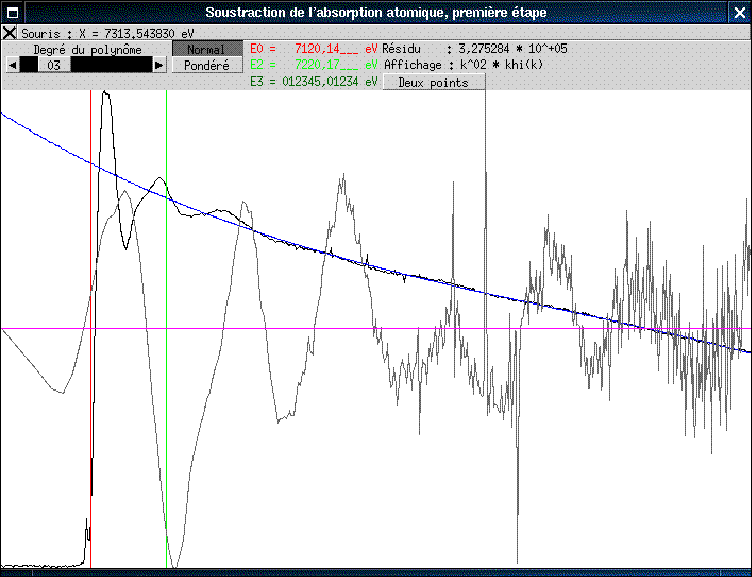
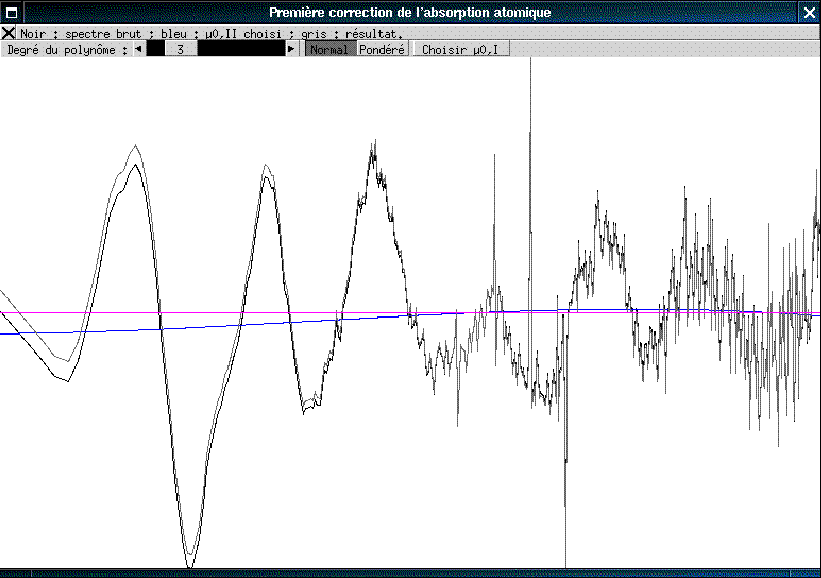

- Les modifications d'un paramètre lors d'une
étape peuvent être automatiquement propagées
aux étapes suivantes (y compris la transformation de
Fourier) ;
- Pondération possible par kn, n
= 0 à 9 ;
- Normalisation soit à l'aide du µ0 extrait, soit
à l'aide des modèles théoriques (Vectoreen,
McKale,...).
Filtrage des spectres
Il se fait de façon classique, en utilisant les
propriétés de la transformation de Fourier.
- Choix libre de la fonction d'apodisation
(fenêtre) parmi plusieurs fonctions de base :
rectangulaire, gaussienne, de Hanning, de Hamming
(symétrique ou non), de Norton-Beer, de Kaiser-Bessel,...
- Paramétrage complet de l'intervalle de calcul et du pas
à utiliser ;
- Correction de phases et amplitudes possible avant calcul de la
TF ;
- Calcul possible soit par un algorithme rapide, soit par calcul
direct des intégrales (donc sans modification
préalable du signal expérimental) ;
- Calcul possible des barres d'erreur sur la partie
réelle et la partie imaginaire de la TF, avec matrice de
covariance correspondante sur demande ;
- Aperçu en temps réel de l'influence de la
fenêtre sur la TF (désactivable pour les machines
lentes) ;
- Paramétrage du filtre dans l'espace des R parmi un
choix de fonction important ;
- La fonction d'apodisation est mémorisée, de
façon à corriger son influence après TF
inverse ;
- Extraction possible des phases et amplitudes du signal lors de
la TF inverse.

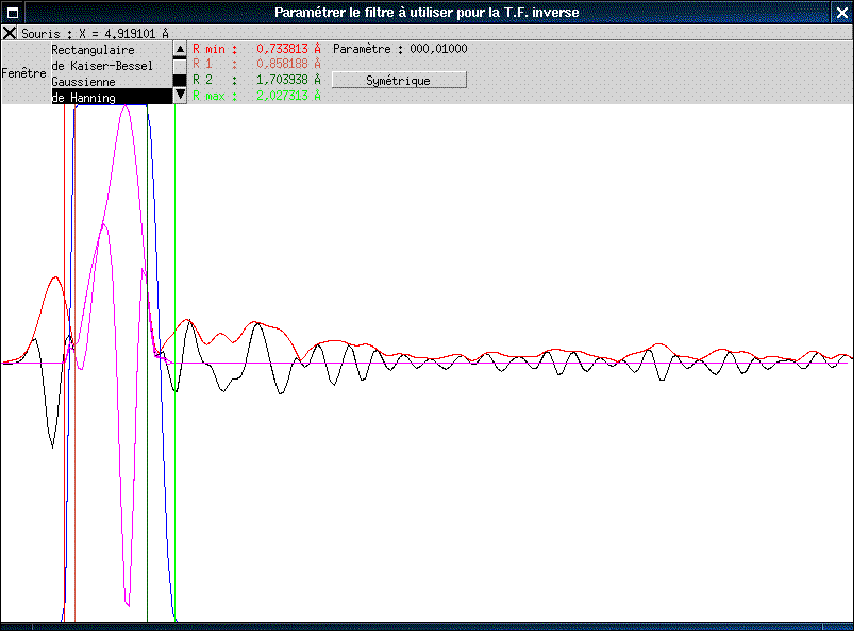
Création d'un modèle
structural
LASE dispose d'un certain nombre d'options permettant la
réalisation facile de modèles structuraux,
nécessaires au calcul des spectres EXAFS théoriques qui
seront utilisés lors de l'étape d'ajustement.
Actuellement, ces modèles structuraux sont utilisables pour la
création de fichiers de paramètres FEFF.
Pour ses modèles structuraux, LASE raisonne en terme de
« molécules ». Une molécule est un
ensemble d'atomes, réunis pour une raison logique :
molécule au sens propre, maille (ou groupe de mailles)
cristallines,... Dans cette molécule, un ou plusieurs atomes
peuvent être définis comme
« absorbeurs » : un absorbeur regroupe un
spectre EXAFS expérimental, éventuellement des spectres
EXAFS modèles, un ensemble de chemins de rétrodiffusion
du photoélectron, ainsi que des informations
nécessaires aux ajustements.
Chargement de données structurales
LASE permet de charger trois types de fichiers structuraux :
les fichiers *.COR créés par la Cambridge
Cristallographic Databank ; les fichiers au format PDB et les
fichiers ASCII créés par Carine. L'utilisation du
logiciel Babel, qui permet de créer des fichiers au format PDB
à partir de nombreux formats de fichiers décrivant des
structures chimiques, permet en pratique de charger dans LASE
à peu près n'importe quelle structure.
Il est aussi possible de créer une structure directement dans LASE, soit ŕ
partir des coordonnées dans un repčre orthonormé, soit ŕ partir des
coordonnées cirstallographiques fractionnaires. Pour quelques groupes d'espace
(P1, P-1, P21, C2/c, Pnma), il est possible de générer automatiquement les
atomes en positions équivalentes ; il est aussi possible de générer plusieurs
mailles en une fois.
Visualisation du modèle structural
À condition de disposer des bibliothèques Open-GL
adéquates (MESA convient
parfaitement), il est possible de visualiser dans LASE le modèle
structural créé soit à partir des fichiers
chargés, soit directement dans le programme, ŕ partir des coordonnées
cristallographiques et du groupe d'espace.
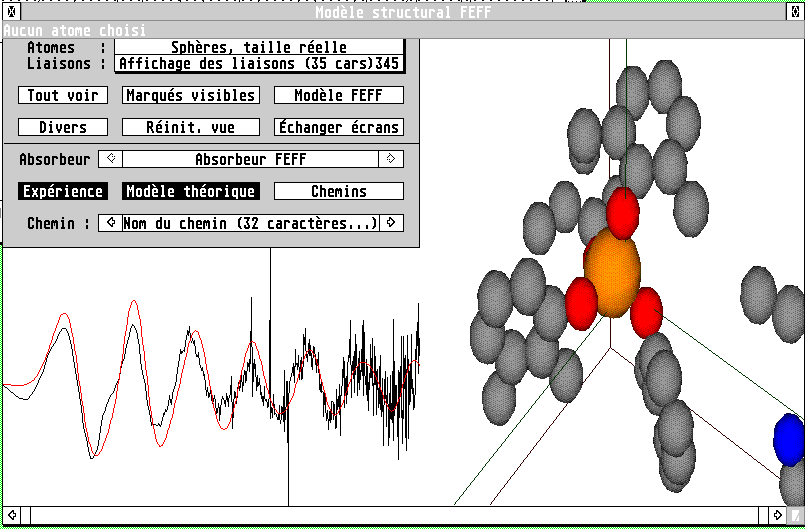
Dans ce cas, il est possible de connaître, directement en cliquant
sur un ou plusieurs atomes, leurs coordonnées, les distances qui les
séparent,... On peut aussi directement ajouter ou enlever des atomes
d'un modèle destiné à FEFF. Si un absorbeur est
sélectionné, les différents spectres EXAFS
associés sont visualisés. Il est aussi possible de voir les
différents chemins de rétrodiffusion, et leur contribution au
signal aprčs ajustement.
Création d'un modèle pour FEFF
LASE dispose d'une interface graphique permettant de
paramétrer toutes les options de FEFF6, à l'exception
de la commande OVERLAP.
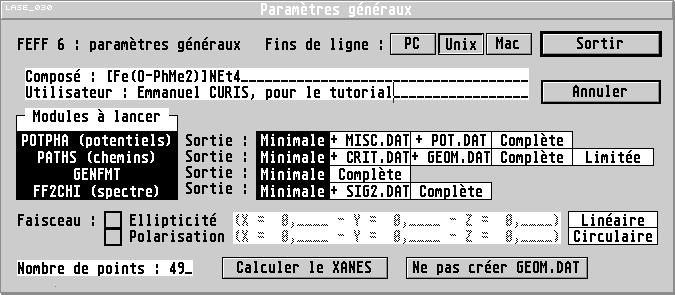


La création d'un modèle structural peut se faire
automatiquement à partir des structures
mémorisées par LASE, soit en intégrant tous les
atomes mémorisés, soit uniquement les atomes
situés à moins d'une certaine distance de l'atome
défini comme absorbeur pour FEFF. Il est aussi possible de
modifier ou de créer des atomes directement depuis l'interface
de paramétrage de FEFF.
Recherche des paramètres
optimaux
LASE permet de réaliser des ajustements dans l'espace des
k (donc sur les spectres EXAFS), en tenant compte ou non des
barres d'erreur déterminées lors de l'extraction du
signal. Cet ajustement repose sur un modèle en chemins de
diffusion dans l'environnement de l'atome absorbeur ; dans le
cas où seule la réflexion simple intervient, un chemin
est équivalent à une couche de réflecteurs.
Pour chaque chemin, il est possible d'ajuster la
dégénérescence (N, nombre de voisins), le
facteur de Debye-Waller, la distance, quatre cumulants
permettant de tenir compte d'une importante anharmonicité des
liaisons, et éventuellement les valeurs de correction de
l'énergie de seuil et de libre parcours moyen.
En plus de ces variables, il est possible d'ajuster une correction
de l'énergie de seuil générale pour le spectre,
ainsi que le facteur de réduction électronique
S02. Dans le cas d'ajustement simultané
sur plusieurs absorbeurs (travail sur plusieurs
éléments ou avec plusieurs environnements pour un
même élément), ces paramètres sont
associés à l'absorbeur. Dans le cas de systèmes
avec plusieurs sites possibles pour le même type d'atome, il
est aussi possible d'ajuster la contribution des différents
sites au spectre total.
Au cours de l'ajustement, le spectre modélisé est
réaffiché en temps réel, afin de suivre la
minimisation à l'écran.
Utilisation des résultats des calculs de FEFF
LASE permet de lire les fichiers paths.dat
créés par FEFF afin de construire un modèle
structural (s'il n'est pas déjà présent en
mémoire), les différents chemins de réflexion
associés et les amplitudes et déphasages
correspondants.
Validation des résultats obtenus
LASE dispose des fonctions nécessaires à la
simulation Monte-Carlo des ajustements, permettant de
déterminer les valeurs moyennes et les erreurs sur ces valeurs
pour les différents paramètres recherchés au
cours de l'ajustement, sans autre hypothèse que le
caractère gaussien de la distribution des valeurs
expérimentales.
Nouveautés
Version du 22 janvier 2001
- la sélection automatique du potentiel de l'atome absorbeur, dans
le paramétrage de FEFF (bouton choisir) est plus
intelligente : elle choisit d'abord parmi les atomes marqués comme
absorbeurs de la molécule courante ; s'il n'y en a pas, parmi
les absorbeurs potentiels de la molécule courante , en dernier
recours, parmi tous les atomes en mémoire.
- la sauvegarde des fichiers feff.inp ne plante plus quand un
potentiel 0 est défini sans atome associé, mais avec des atomes pour
les autres potentiels (je ne pense pas qu'un tel fichier soit accepté
par FEFF, ceci dit).
- Version X-Window : il est possible, dans les champs
éditables (textes), de passer en mode « remplacement »
(curseur carré, ancien mode) ou « insertion » (curseur
linéaire), en utilisant la touche Inser
- Version Windows : recompilée avec la nouvelle
cygwin, ce qui permet le chargement correct des fichiers
expérimentaux du poste expérimental exafs-2. Une série de
redimensionnements des fenętres ne conduit plus ŕ un écran
blanc. Cette version reste expérimentale ! Si
vous le pouvez, utilisez la version unix.
Version du 12 janvier 2001
- possibilité de calculer l'angle entre trois atomes du modčle
structural, en cliquant sur ces trois atomes sur la vue 3D
- calcul des distances entre atomes en cliquant sur les divers
atomes sur la vue en 3D, selon plusieurs modes (en particulier, en
choisissant un atome de référence et en calculant toutes les
distances par rapport ŕ cet atome)
- possibilité de créer et sélectionner les atomes absorbeurs
directement sur la vue tridimensionnelle (Ctrl-clic sur l'atome
choisi)
- possibilité de scinder en plusieurs zones la fenętre d'affichage
des spectres superposés, les spectres et grossissements affichés dans
chaque zone étant indépendants et librement définissables (on peut
ainsi avoir ŕ gauche la comparaison des spectres exafs, ŕ droite
celle des transformées de Fourier dans la męme fenętre)
- possibilité d'adapter le grossissement en Y pour un intervalle
en X défini par l'utilisateur
- possibilité d'afficher, dans la fenętre de normalisation d'un
seuil, un seuil de référence pour faciliter la normalisation relative
de deux seuils (appuyer sur r dans la fenętre)
Version d'aoűt 2000
- corrigé une erreur dans la génération des spectres ŕ ajuster par
la méthode de Monte-Carlo, pouvant conduire ŕ sous-estimer les
incertitudes sur les paramčtres ajustés dans certains cas.
Version du 14 juillet 2000
- compile proprement sur Irix 5 et Irix 6.2 ;
- plus de « BadMatch » en ouvrant la fenętre d'affichage 3D sous
Irix avec un écran en 256 couleurs ;
- plus d'erreur en corrigeant les énergies avec une énergie nulle.
- possibilité de ne sélectionner que certains spectres d'un fichier męme
quand on ne regroupe pas ces spectres en un seul spectre moyen.
Version du 24 mai 2000
- possibilité de sauvegarder et charger les paramčtres d'extraction des
oscillations EXAFS (fichier ASCII qui peut ętre lu par n'importe quelle
version de LASE, sur toute machine) ;
- possibilité de déplacer un atome en modifiant uniquement sa distance ŕ
l'absorbeur, en le déplaçant le long de la droite (absorbeur, atome) ;
- génération des fichiers POV, pour réaliser des illustrations ŕ partir de
la vue tridimensionnelle, améliorée : elle tient compte des paramčtres
d'affichage (taille des sphčres, sélection d'atomes visibles) et inclus le
chemin de réflexion sélectionné, s'il existe ;
- corrigé la génération des fichiers FEFF (type de potentiel ŕ utiliser,
auparavant incorrectement indiqué) ;
- paramčtres statistiques d'un graphe : estime maintenant la médiane et le
moment centré absolu d'ordre 1 ;
- possibilité de calculer la contribution individuelle de chaque couche
utilisée dans un ajustement. Cette contribution apparaît sur la vue 3D quand
le chemin correspondant est sélectionné, pour une comparaison rapide au
signal expérimental ;
- les paramčtres d'extraction de µ0, I quand on tronque la
fin du spectre (utilisation d'un 2e point de contrôle marquant la
fin) sont adaptés dans la boîte de dialogue de reproduction de
l'extraction ;
- groupes d'espace reconnus : P1, P-1, P2, P2/c, C2/c, Pnma.
Pour davantage d'informations
N'hésitez pas à me contacter par courrier
électronique :
Emmanuel CURIS